1 拼音
GB 30615—2014 shí pǐn tiān jiā jì zhú yè kàng yǎng huà wù
中华人民共和国国家标准 食品安全国家标准 GB 30615—2014《食品添加剂 竹叶抗氧化物》由中华人民共和国国家卫生和计划生育委员会于2014年4月29日发布,自2014年11月1日起实施。
食品安全国家标准
2 1 范围
本标准适用于以刚竹属 (Phyllostachys Siet. Et Zucc) 竹种的叶为原料,经提取、精制而成的食品添加剂竹叶抗氧化物。
注: 用于生产食品添加剂竹叶抗氧化物的竹叶原料为被子植物门(Angiospermae)、单子叶植物纲(Monocotyledonae)、禾本目(Graminales)、禾本科(Graminae)、竹亚科(Bambusoideae)、刚竹属(Phyllostachys)品种 1~2 年生的叶子。
3 2 分类
竹叶抗氧化物根据其溶解性分为水溶性产品和脂溶性产品。其水溶性产品的主要有效成分为竹叶碳苷黄酮(异荭草苷、荭草苷、牡荆苷、异牡荆苷)和对香豆酸、绿原酸等;脂溶性产品的主要有效成分为对香豆酸、阿魏酸、苜蓿素以及竹叶黄酮的酯化产物等。
4 3 主要成分的化学名称、结构式、分子式和相对分子质量
4.1 3.1 异荭草苷
4.1.1 3.1.1 化学名称
4.1.2 3.1.2 分子式
C21H20O11
4.1.3 3.1.3 结构式

4.1.4 3.1.4 相对分子质量
448.38(按2007年国际相对原子质量)
4.2 3.2 对香豆酸
4.2.1 3.2.1 化学名称
4.2.2 3.2.2 分子式
C9H8O3
4.2.3 3.2.3 结构式

4.2.4 3.2.4 相对分子质量
164.16(按2007年国际相对原子质量)

6 附录A 检验方法
6.1 A.1 安全提示
本标准试验方法中使用的部分试剂具有毒性或腐蚀性,按相关规定操作,操作时需小心谨慎。若溅到皮肤上应立即用水冲洗,严重者应立即治疗。在使用挥发性酸时,要在通风柜中进行。
6.2 A.2 一般规定
本标准所用试剂和水,在没有注明其他要求时,均指分析纯试剂和GB/T 6682规定的三级水。试验中所用标准溶液、杂质标准溶液、制剂及制品, 在没有注明其他要求时,均按GB/T 601、 GB/T 602、 GB/T 603的规定制备。实验中所用溶液在未注明用何种溶剂配制时,均指水溶液。
6.3 A.3 鉴别试验
6.3.1 A.3.1 化学试剂鉴别
6.3.1.1 A.3.1.1 试剂和材料
6.3.1.2 A.3.1.2 鉴别方法
A.3.1.2.1 称取约0.5 g 试样溶于100 mL乙醇中, 取此溶液1 mL滴加2~3滴三氯化铁乙醇溶液,溶液显深蓝色或蓝紫色。
A.3.1.2.2 称取约0.5 g 试样溶于100 mL乙醇中,取此溶液1 mL滴加2~3滴三氯化铝乙醇溶液,溶液显鲜黄色。
6.4 A.3.2 光谱学鉴别
6.4.1.1 A.3.2.1 红外透射光谱鉴别
采用溴化钾压片法,按照GB/T 6040 的规定进行试验,试样的红外光谱应与对照图谱一致(对照图谱见附录B)。
6.4.1.2 A.3.2.2 紫外吸收光谱鉴别
称取约15 mg试样溶于100 mL色谱纯甲醇中,在200 nm~600 nm波长范围内进行紫外扫描,试样的紫外光谱应与对照图谱一致(对照图谱见附录C)。
6.4.2 A.3.3 有效成分分析及其指纹图谱
6.4.2.1 A.3.3.1 方法提要
试样用色谱纯甲醇溶解,以乙腈-乙酸水溶液(1+99)为流动相,用以C18 为填料的液相色谱柱和紫外检测器或二级管阵列检测器,用含异荭草苷、荭草苷、异牡荆苷、牡荆苷、对香豆酸、绿原酸、咖啡酸、阿魏酸等八个对照品的标准图谱和试样图谱进行对照,确定竹叶抗氧化物产品的有效成分及类别差异。
6.4.2.2 A.3.3.2 试剂和材料
A.3.3.2.1 乙腈(色谱纯)。
A.3.3.2.2 甲醇(色谱纯)。
A.3.3.2.3 冰乙酸(优级纯)。
A.3.3.2.4 水(超纯水)。
A.3.3.2.5 异荭草苷、荭草苷、异牡荆苷、牡荆苷、对香豆酸、绿原酸、咖啡酸、阿魏酸标准品: 纯度 ≥98.0%。
A.3.3.2.6 混标溶液:分别称取上述8个对照品(A.3.3.2.5)各5 mg(精确至0.0001 g) ,用甲醇溶解并定容至10 mL,混匀,置冰箱中保存,此溶液为八个对照品的混标溶液。
6.4.2.3 A.3.3.3 仪器和设备
A.3.3.3.1 高效液相色谱仪。
A.3.3.3.2 紫外光检测器:可变波长。
A.3.3.3.3 数据处理系统:色谱工作站或数据处理机。
6.4.2.4 A.3.3.4 参考色谱条件
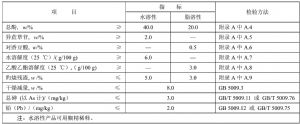
推荐的色谱柱及典型操作条件见表A.1,竹叶抗氧化物指纹图谱测定典型高效液相色谱图见附录D中D.1 和D.2。其他能达到同等分离程度的色谱柱和色谱操作条件均可使用。
表A.1 色谱柱和典型色谱操作条件
6.4.2.5 A.3.3.5 分析步骤
A.3.3.5.1 试样处理
准确称取试样100 mg (精确至0.000 1 g) ,用甲醇溶解并定容至100 mL,经微孔滤膜(0.45 µm)过滤,即得试样溶液。
A.3.3.5.2 试样测定
准确吸取试样溶液10 μL,在规定色谱条件下,进行色谱分析,以保留时间定性。试样的高效液相色谱图应与对照图谱一致(对照图谱见附录D)。
6.5 A.4 总酚测定
6.5.1 A.4.1 方法提要
福林(Folin)试剂在碱性条件下极不稳定,可使酚类化合物还原而呈蓝色反应。在对羟基苯甲酸浓度0.0005 mg/mL~0.016 mg/mL范围内,其浓度与吸光度符合比尔定律,可用对羟基苯甲酸为对照品进行定量比色测定得试样的总酚含量。
6.5.2 A.4.2 试剂和材料
A.4.2.3 碳酸钠溶液:200 g/L。
A.4.2.5 对羟基苯甲酸标准溶液: 0.250 mg/mL, 称取干燥至恒重的对羟基苯甲酸标准品25.0 mg,用乙醇水溶液(A.4.2.1)溶解并定容至100 mL。
A.4.2.6 福林试剂(自行配制或外购) :于2000 mL磨口烧瓶中放入100 g钨酸钠(Na2WO4•2H2O) 、25g钼酸钠(Na2MoO4•2H2O) 、700 mL水、50 mL 85%磷酸(H3PO4) 、100 mL盐酸,小火沸腾回流10 h,去除冷凝器后,加入50 g硫酸锂 (Li2SO4) 、 50 mL水和数滴溴水(99%), 摇匀,在通风柜内进行,再煮沸15 min以去除过量的溴,冷却后加水定容至1000 mL,混合均匀,过滤。试剂呈金黄色,储存于棕色瓶内,使用时以1份原液于2份水稀释均匀。
6.5.3 A.4.3 仪器和设备
A.4.3.1 分光光度计。
A.4.3.2 万分之一电子天平:感量 0.0001 g。
A.4.3.3 移液枪(或移液管)。
A.4.3.4 容量瓶。
6.5.4 A.4.4 测定步骤
6.5.4.1 A.4.4.1 试样处理
准确称取竹叶抗氧化物试样100 mg(精确至0.0001 g),水溶性产品用乙醇水溶液(A.4.2.1)溶解并定容至100 mL,脂溶性产品用少量乙醇水溶液 (A.4.2.2) 溶解后、再用乙醇水溶液 (A.4.2.1) 定容至100 mL;用定性滤纸过滤,即得试样溶液。
6.5.4.2 A.4.4.2 标准曲线的绘制
准确吸取对羟基苯甲酸标准溶液0 mL、0.05 mL、0.10 mL、0.20 mL、0.40 mL、0.80 mL、1.20 mL,相当于对羟基苯甲酸0 mg、0.0125 mg、0.025 mg、0.050 mg、0.10 mg、0.20 mg、0.30 mg 移入25 mL 具塞试管中,分别用水稀释至10.0 mL;各加入1.0 mL 福林试剂和2.0 mL 20%碳酸钠溶液,试管在50 ℃±1 ℃的恒温水浴中加热30 min,然后用水冷却并稀释至25 mL。室温放置30 min,用1 cm比色杯测定745 nm的吸光度。以吸收度为纵坐标、浓度为横坐标绘制标准曲线或求取线性回归方程。
6.5.4.3 A.4.4.3 试样测定
准确吸取试样溶液1 mL,按上述标准曲线制作中的操作步骤(A.4.4.2)于745 nm处进行吸光度测定。
6.5.5 A.4.5 结果计算
总酚(以对羟基苯甲酸计)的质量分数w1 按公式(A.1)计算:

式中:
m1——依据标准曲线计算出的待测液中总酚含量的数值,单位为毫克(mg);
实验结果以平行测定结果的算术平均值为准(保留一位小数)。在重复性条件下获得的两次独立测定
结果的相对差值不超过 5.0%。
6.6 A.5 异荭草苷测定
6.6.1 A.5.1 方法提要
试样经甲醇溶解,以乙腈-乙酸水溶液(1+99)为流动相,用以C18 为填料的液相色谱柱和紫外检测器或二极管阵列检测器,对试样中的异荭草苷进行反相高效液相色谱分离和测定,与标准品保留时间比较定性,峰面积外标法定量。
6.6.2 A.5.2 试剂和材料
A.5.2.1 乙腈(色谱纯)。
A.5.2.2 甲醇(色谱纯)。
A.5.2.3 冰乙酸(优级纯)。
A.5.2.4 水(超纯水)。
A.5.2.6 异荭草苷标准溶液:准确称取异荭草苷标准品5 mg(精确至0.000 1 g),用甲醇溶解并定容至10mL,混匀,置冰箱中保存。此溶液1 mL含异荭草苷0.5 mg。
6.6.3 A.5.3 仪器和设备
A.5.3.1 高效液相色谱仪。
A.5.3.2 紫外光检测器:可变波长。
A.5.3.3 数据处理系统:色谱工作站或数据处理机。
6.6.4 A.5.4 参考色谱条件
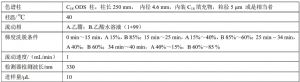
推荐的色谱柱及典型操作条件见表A.2,其他能达到同等分离程度的色谱柱和色谱操作条件均可使用。
表A.2 色谱柱和典型色谱操作条件

6.6.5 A.5.5 分析步骤
6.6.5.1 A.5.5.1 试样处理
准确称取竹叶抗氧化物试样100 mg (精确至0.0001 g) ,用甲醇溶解并定容至100 mL,经微孔滤膜(0.45µm)过滤,即得试样溶液。
6.6.5.2 A.5.5.2 标准曲线的绘制
准确吸取一定量的异荭草苷标准溶液,分别稀释4、8、10、16、20 和40 倍,进样10 μL,在规定色谱条件下,进行色谱分析。 根据异荭草苷标准溶液的不同进样量及相应谱峰面积,以色谱峰面积为纵坐标、异荭草苷浓度为横坐标,绘制标准曲线。
6.6.5.3 A.5.5.3 试样测定
准确吸取试样溶液10 μL,在规定色谱条件下,进行色谱分析,以保留时间定性,峰面积外标法定量。
6.6.6 A.5.6 结果计算
异荭草苷的质量分数w2 按公式(A.2)计算:

式中:
c1——依据标准曲线计算出的待测液中的异荭草苷浓度的数值,单位为毫克每毫升(mg/mL);
实验结果以平行测定结果的算术平均值为准(保留一位小数)。在重复性条件下获得的两次独立测定结果的相对差值不超过 2.5%。
6.7 A.6 对香豆酸测定
6.7.1 A.6.1 方法提要
试样经甲醇溶解,以乙腈-乙酸水溶液(1+99)为流动相,用以C18 为填料的液相色谱柱和紫外检测器或二极管阵列检测器,对试样中的对香豆酸进行反相高效液相色谱分离和测定,与标准品保留时间比较定性,峰面积外标法定量。
6.7.2 A.6.2 试剂和材料
A.6.2.1 乙腈(色谱纯)。
A.6.2.2 甲醇(色谱纯)。
A.6.2.3 冰乙酸(优级纯)。
A.6.2.4 水(超纯水)。
A.6.2.5 对香豆酸标准品:纯度≥ 98.0%。
A.6.2.6 对香豆酸标准溶液:准确称取对香豆酸标准品10 mg(精确至0.0001 g),用甲醇溶解并定容至10mL,混匀,置冰箱中保存。此溶液1 mL含对香豆酸1.0 mg。
6.7.3 A.6.3 仪器和设备
A.6.3.1 高效液相色谱仪。
A.6.3.2 紫外光检测器:可变波长。
A.6.3.3 数据处理系统:色谱工作站或数据处理机。
6.7.4 A.6.4 参考色谱条件
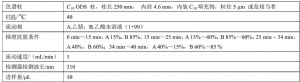
推荐的色谱柱及典型操作条件见表A.3,其他能达到同等分离程度的色谱柱和色谱操作条件均可使用。
表A.3 色谱柱和典型色谱操作条件
6.7.5 A.6.5 分析步骤
6.7.5.1 A.6.5.1 试样处理
准确称取竹叶抗氧化物试样100 mg (精确至0.0001 g) ,用甲醇溶解并定容至100 mL,经微孔滤膜(0.45µm)过滤,即得试样溶液。
6.7.5.2 A.6.5.2 标准曲线的绘制
准确吸取一定量的对香豆酸标准溶液,分别稀释8、10、16、32、64、128 和256 倍,进样10 μL,在规定色谱条件下,进行色谱分析,根据对香豆酸标准溶液的不同进样量及相应谱峰面积,以色谱峰面积为纵坐标、对香豆酸浓度为横坐标,绘制标准曲线。
6.7.5.3 A.6.5.3 试样测定
准确吸取试样溶液10 μL,在规定色谱条件下,进行色谱分析,以保留时间定性,峰面积外标法定量。
6.7.6 A.6.6 结果计算
对香豆酸的质量分数w3按公式(A.3)计算:

式中:
c2——依据标准曲线计算出的待测液中对香豆酸浓度的数值,单位为毫克每毫升(mg/mL);
实验结果以平行测定结果的算术平均值为准(保留一位小数)。在重复性条件下获得的两次独立测定结果的相对差值不超过 2.5%。
6.8 A.7 水溶解度测定
6.8.1 A.7.1 仪器和设备
A.7.1.1 电热恒温干燥箱。
A.7.1.2 百分之一电子天平:感量 0.01 g。
6.8.2 A.7.2 分析步骤
取200 mL 的具塞锥形瓶,干燥至恒重(m5)后,准确称取100 g蒸馏水(精确至0.01 g),然后加入6 g~10 g水溶性竹叶抗氧化物(精确至0.01 g),在25 ℃条件下边加边搅拌,在5 min内使其充分溶解,静置10 min,倒出上清液,瓶及残渣干燥至恒重(m7),根据减轻的质量计算水溶解度。
6.8.3 A.7.3 结果计算
水溶解度以w4 表示,其数值以克每百克(g/100 g)表示,按公式(A.4)计算:

式中:
实验结果以平行测定结果的算术平均值为准(保留一位小数)。在重复性条件下获得的两次独立测定结果的相对差值不超过 5.0%。
6.9 A.8 乙酸乙酯溶解度测定
6.9.1 A.8.1 试剂和材料
乙酸乙酯。
6.9.2 A.8.2 仪器和设备
A.8.2.1 电热恒温干燥箱。
A.8.2.2 百分之一电子天平:感量 0.01 g。
6.9.3 A.8.3 分析步骤
取200 mL的具塞锥形瓶,干燥至恒重(m8)后,准确称取100 g乙酸乙酯(精确至0.01 g),然后加入5g~10 g脂溶性竹叶抗氧化物(精确至0.01 g),在25 ℃条件下边加边搅拌,在5 min内使其充分溶解,静置10 min,倒出上清液,瓶及残渣干燥至恒重(m10),根据减轻的质量计算乙酸乙酯溶解度。
6.9.4 A.8.4 结果计算
乙酸乙酯溶解度以w5 表示,其数值以克每百克(g/100 g)表示,按公式(A.5)计算:

式中:
实验结果以平行测定结果的算术平均值为准(保留一位小数)。在重复性条件下获得的两次独立测定结果的相对差值不超过 5.0%。
6.10 A.9 灼烧残渣测定
6.10.1 A.9.1 分析步骤
称取约2 g 试样(精确至0.0001 g),置于预先恒重的坩埚内,先用小火缓缓加热至完全炭化,然后小心移入高温炉内于600 ℃±25 ℃灼烧至恒重。
6.10.2 A.9.2 结果计算

式中:
m12——坩埚质量的数值,单位为克(g);
实验结果以平行测定结果的算术平均值为准(保留一位小数)。在重复性条件下获得的两次独立测定结果的相对差值不超过 5.0%。
7 附录B 竹叶抗氧化物红外透射光谱图
7.1 B.1 水溶性竹叶抗氧化物红外透射光谱示意图
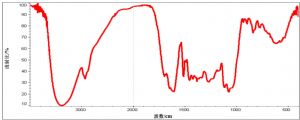
7.2 B.2 脂溶性竹叶抗氧化物红外透射光谱示意图
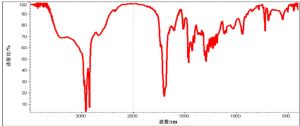
注:水溶性竹叶抗氧化物在3400 cm-1、2930 cm-1、1610 cm-1、1075 cm-1等附近有特征性吸收,分别示酚羟基、苯环上碳氢键、苯环骨架及羰基的伸缩振动;脂溶性竹叶抗氧化物在3400 cm-1左右的酚羟基峰大大减弱,1700 cm-1附近出现强的羰基吸收峰。
8 附录C 竹叶抗氧化物紫外吸收光谱图
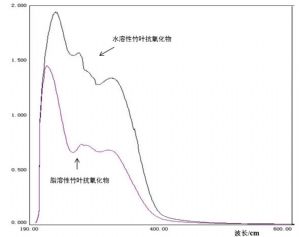
注:竹叶抗氧化物的紫外光谱图在240 nm~280 nm之间有一强吸收峰,在300 nm~350 nm之间有一强吸收峰。同浓度下水溶性产品的紫外吸收強度大于脂溶性产品。

